
Beta-Thalassemia
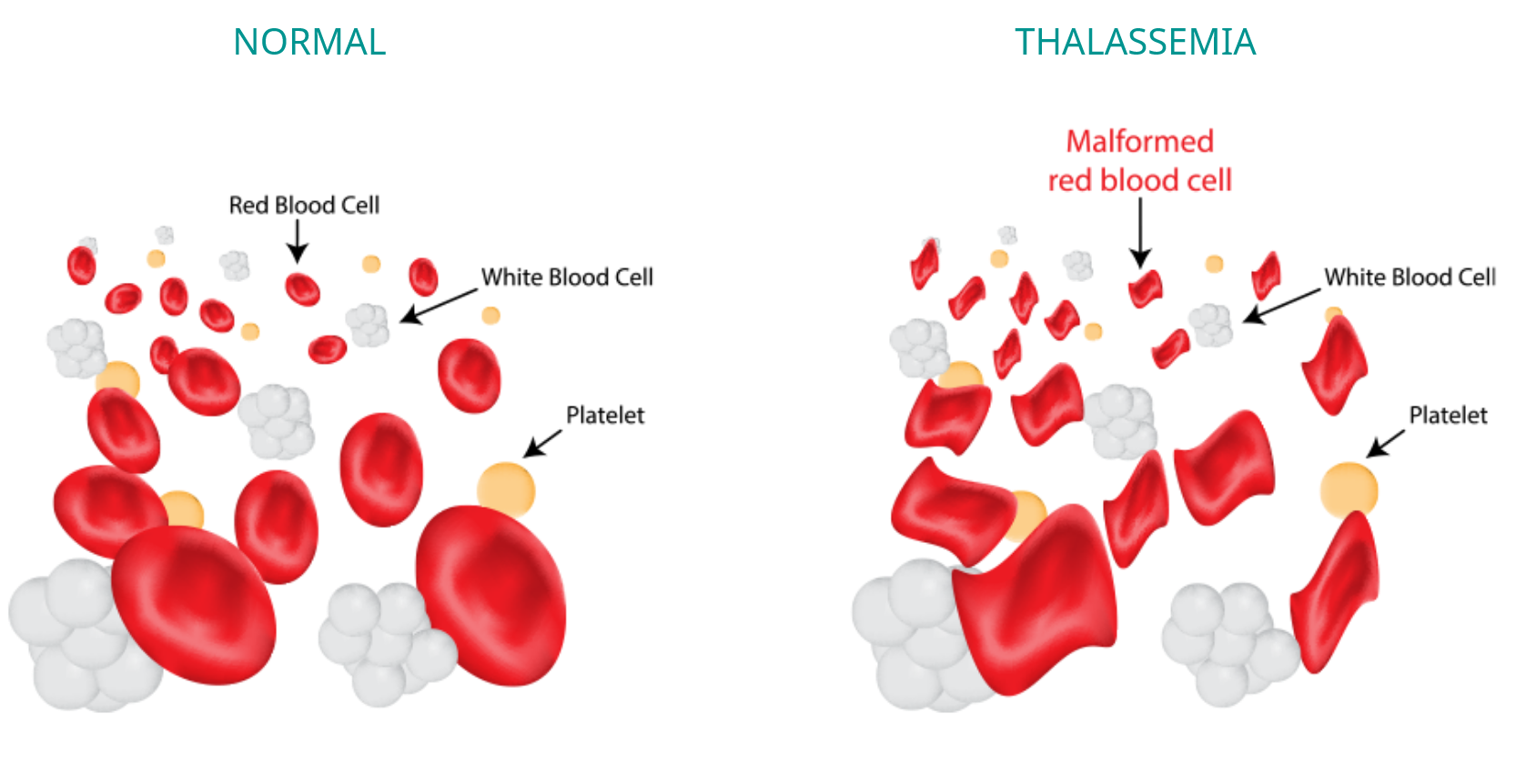
Beta-thalassemia is a blood disorder caused by mutations in the HBB gene, resulting in red blood cells that are microcytic and do not contain enough functional hemoglobin.1,2 Low levels of hemoglobin lead to a lack of oxygen in many parts of the body.1
This is an autosomal recessive disease, meaning the parents are carriers of the disease (but are typically asymptomatic), and the affected individual has two copies of the HBB gene with pathogenic mutations.1
In addition to low levels of hemoglobin, people with Beta-thalassemia also have anemia, resulting in pale skin, weakness, fatigue, and serious complications. Furthermore, there is an increased risk of developing abnormal blood clots.1
Beta-thalassemia is relatively rare in the United States, but is common in other parts of the world. The incidence of symptomatic cases is estimated to be approximately 1 in 100,000 individuals in the general population.3
- Genetics Home Reference. National Institutes of Health. https://ghr.nlm.nih.gov/condition/beta-thalassemia. Last accessed: December 19, 2019.
- Origa R. 2000 Sep 28 [Updated 2018 Jan 25]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1426/. Last accessed: December 20, 2019.
- National Organization for Rare Disorders (NORD). https://rarediseases.org/rare-diseases/thalassemia-major/. Last accessed: December 20, 2019.