Welcome to Chiesi
USA
Home page > Fabry Disease

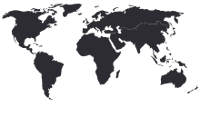
Fabry Disease
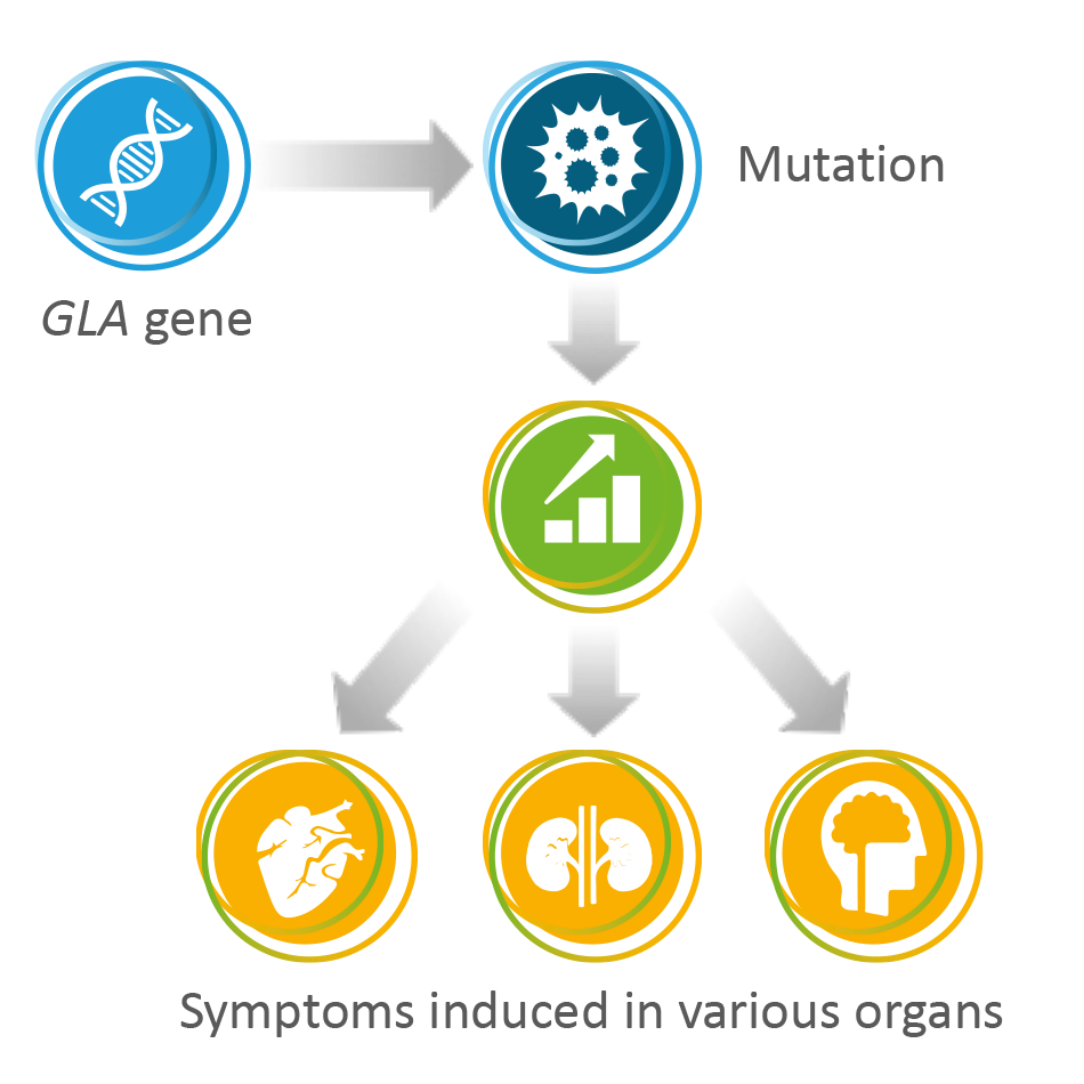
Fabry disease is a lysosomal storage disorder, meaning that a glycosphingolipid called GL-3 accumulates in the lysosomes, causing tissue damage; many cell types are affected.1
The disease is caused by mutations in the GLA gene, resulting in nonfunctional or dysfunctional alpha-galactosidase A, a lysosomal enzyme. The mutations can be inherited, so multiple family members can have the disease.1
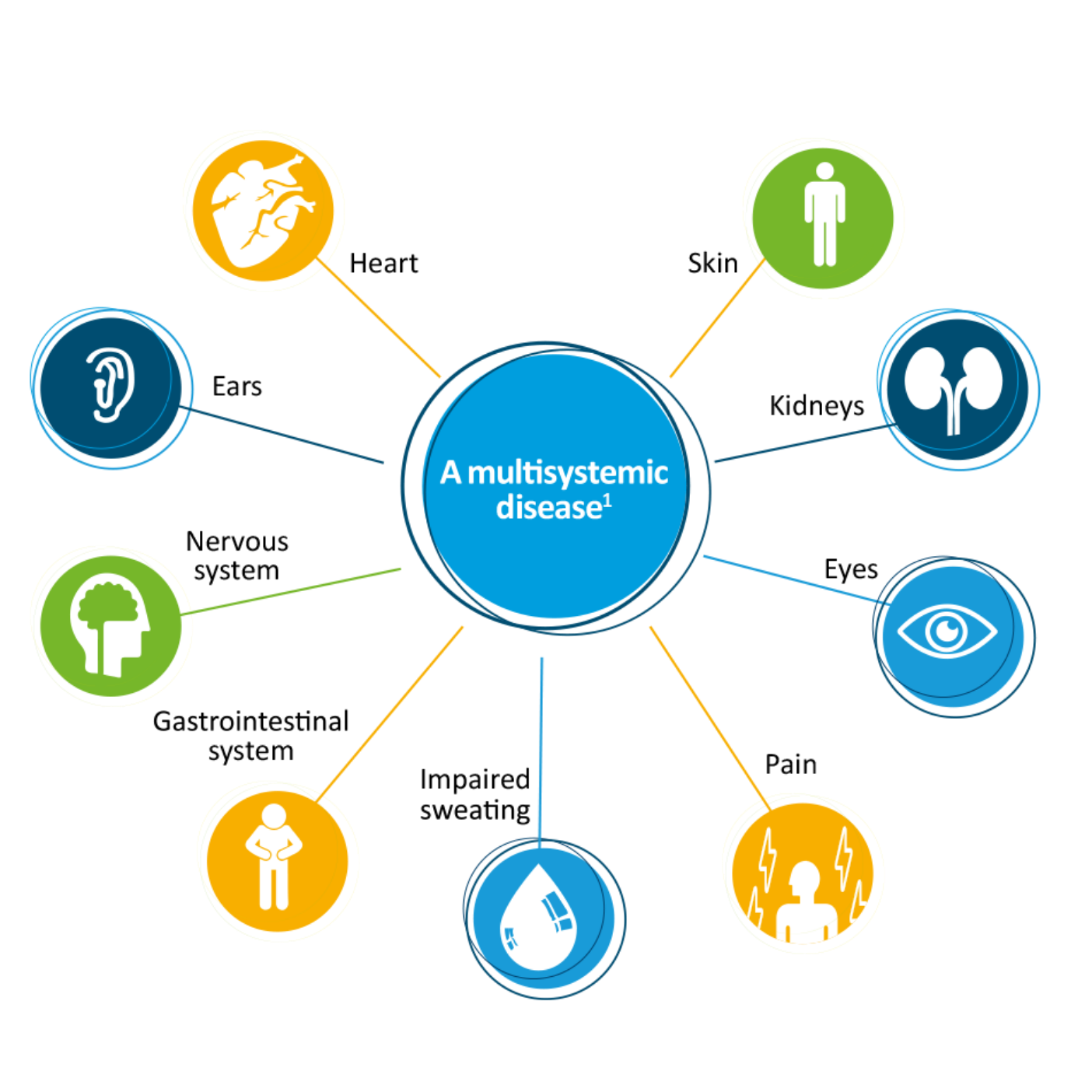
Fabry disease is a multisystemic disease, affecting many organs, including the heart, kidney and nervous system, resulting in life-threatening complications and a reduced life expectancy. Early signs of the disease start in childhood and adolescence, but it is a progressive, lifelong condition.1,2
Newborn screening has now been performed in several countries, yielding a prevalence ranging from 1 in 1,368 to 1 in 8,882 births.2
- Wanner C, et al. Mol Genet Metab 2018;124(3):189-203.
- Cairns T, et al. Postgrad Med J 2018;94(1118):709-713.